Asymetrická hydrogenace
Asymetrická hydrogenace je chemická reakce, při které se dva atomy vodíku navážou na molekulu substrátu s trojrozměrnou prostorovou selektivitou. Příčina této selektivity by neměla pocházet ze samotné molekuly substrátu, ale z jiných reaktantů nebo z katalyzátoru. Tímto se může chiralita přenést z reaktantu na substrát, čímž vznikne produkt obsahující pouze jeden enantiomer. Chiralitu obvykle zajišťuje katalyzátor, díky čemuž jedna molekula může přenést informaci o chiralitě na mnoho molekul substrátu, čímž se množství přítomné chirality navyšuje. Podobné procesy probíhají i v přírodě, kde chirální molekuly, jako jsou některé enzymy, katalyzují tvorbu chirálních center a produktů, například aminokyselin v jednom enantiomeru, který je potřebný ke správné funkci buňky. Napodobením těchto dějů lze připravit mnoho nových syntetických sloučenin interagujících s biologickými systémy konkrétním způsobem, například při vývoji léčiv a agrochemikálií. O rozvoj asymetrické hydrogenace ve výzkumu i průmyslu se zasloužili William Standish Knowles a Rjódži Nojori, kteří v roce 2001 společně obdrželi Nobelovu cenu za chemii.
Historie
editovatV roce 1956 bylo zjištěno, že heterogenní katalyzátor tvořený palladiem na vrstvě hedvábí vyvolává asymetrickou hydrogenaci.[1]
Roku 1968 skupiny, které vedli William Standish Knowles a Leopold Horner, nezávisle na sobě popsaly případy asymetrické hydrogenace pomocí homogenních katalyzátorů. Přestože dosažené enantiomerní přebytky nebyly vysoké, tak tyto reakce ukázaly možnou využitelnost. Do roku 1972 byly dosaženy enantiomení přebytky až 90 % a tímto postupem se začalo vyrábět léčivo na Parkinsonovu nemoc nazývané L-DOPA.[2][3]
.svg)
Rozvoj asymetrické hydrogenace pokračoval několika úspěchy. Henri Kagan vyvinul DIOP, snadno připravitelný C2-symetrický difosfin, který u určitých reakcí poskytoval vysoké enantiomerní přebytky. Rjódži Nojori zavedl pro asymetrické hydrogenace polárních substrátů, jako jsou ketony a aldehydy , katalyzátory založené na rutheniu. Použití P,N ligandů, jež rozšířily spektrum použitelných C2-symetrických ligandů, i když nemají výrazné výhody oproti chirálním ligandům bez rotační symetrie.[4]
V současnosti jsou asymetrické hydrogenace běžné v laboratořích i chemickém průmyslu.
Význam asymetrické hydrogenace byl oceněn v roce 2001 Nobelovou cenou za chemii. kterou získali William Standish Knowles a Rjódži Nojori.
Mechanismus
editovatMechanismy ve vnější sféře
editovatByly navrženy dva hlavní mechanismy asymetrické hydrogenace pomocí komplexů rhodia: nenasycený a dihydridový. Určení konkrétního mechanismu je obtížné, ovšem jejich rozlišení není pro asymetrické hydrogenace příliš důležité, protože vedou ke vzniku stejného meziproduktu ještě předtím, než se stereochemie přesune na molekulu produktu.[5]

Převaha tvorby jednoho enantiomeru oproti druhému se u těchto reakcí většinou vysvětluje sterickými interakcemi mezi ligandem a prochirálním substrátem. S uvažováním těchto interakcí byly vyvinuty kvadrantové diagramy, ve kterých jsou „blokované oblasti“ obarvené šedě, zatímco „otevřené“ obarvené nejsou. U modelované reakce mají větší skupiny na přicházejícím alkenu snahu orientovat se k otevřeným částem diagramu, zatímco menší skupiny putují do blokovaných míst, následně je vodík dodán na zadní stranu alkenu, čímž se nastaví stereochemie. Pro názornost je zobrazena jen jedna strana chirálního fosfinu.

Použité kovy
editovatPlatinové kovy
editovatPrvním kovem použitým k homogenní katalýze asymetrické hydrogenace bylo rhodium,[6] jež je stále široce využívané. Substráty asymetrických hydrogenací s rhodiovými katalyzátory obvykle vyžadují koordinující skupinu v blízkosti alkenu.[5] Tento požadavek je omezující, příslušné skupiny se ovšem vyskytují u mnoha různých sloučenin, jako jsou mimo jiné nenasycené amidy.[7]
Nojoriovy asymetrické hydrogenace se katalyzují rutheniem.[8][9]
Následující práce přinesly rozšíření využití Nojoriova vzorového katalyzátoru, díky čemuž byly zpracovány i tradičně obtížné substráty, jako jsou t-butylketony[10] a 1-tetralony[11] při použití rutheniových katalyzátorů. Přenosová hydrogenace založená na Ru a TsDPEN se také stala komerčně úspěšnou.[12]
Iridiové katalyzátory jsou vhodné pro řadu „netradičních“ substrátů, pro které nebyly nalezeny vhodné katalyzátory založené na Ru nebo Rh.[13] Typickým případem jsou nefunkcionalizované alkeny,[14], jsou však popsány i jiné příklady, jako například ketony.[15][16]
Obvyklou potíží u iridiových katalyzátorů je jejich náchylnost k trimerizaci v roztocích.[16] Použití nekoordinujícího aniontu BAr F
4 − se ukázalo jako nejšířeji uplatnitelné řešení tohoto problému.[16][17] Další postupy sloužící ke zlepšení stability katalyzátorů jsou například přidání další koordinující skupiny na chirální ligand,[15] posílení sterických efektů,[18] použití dendrimerního ligandu,[19] zvýšení „tuhosti“ ligandu,[20] imobilizace ligandu[21] a použití dvou různých kovů (přičemž je jedním z nich iridium).[21]
Zásadité kovy
editovatŽelezo je často používaným kovem při výzkumu katalytických reakcí, protože je oproti ostatním přechodným kovům levnější a méně toxické.[22]
Bylo provedeno několik asymetrických hydrogenací využívajících železo, ovšem rychlosti reakcí i selektivita byly horší než u katalyzátorů založených na ušlechtilých kovech.[23] V některých případech lze použít nanočástice jako aktivní složku in situ, přičemž výsledná dobrá selektivita může být způsobena neřízenou geometrií produktů.[24]
Druhy ligandů
editovatFosfinové ligandy
editovatChirální fosfinové ligandy, obzvláště C2-symetrické, jsou zdrojem chirality u většiny katalyzátorů asymetrických hydrogenací. Patří sem například BINAP, za jehož využití při Nojoriově asymetrické hydrogenaci byla udělena Nobelova cena.[2]
Chiální fosfinové ligandy lze rozdělit na monodentátní a bidentátní a dále podle polohy stereogenního centra – zda se nachází na fosforu, nebo na organických substituentech.
Obzvláště oblíbené jsou ligandy vykazující C2 symetrii, částečně proto, že jsou u nich výrazně omezeny vazebné konformace mezi substrátem a komplexem kov-ligand (což někdy vede k velmi vysoké enantioselektivitě).[25]
Monodentátní fosfiny
editovatMonofosfinové ligandy patří k prvním, které byly použity v asymetrických hydrogenacích; jako příklad lze uvést CAMP.[26]
Při dalším výzkumu tohoto druhu ligandů byly objeveny ligandy s vazbami P-alkyl i P-heteroatom, přičemž ty s vazbami P-heteroatom dávaly, podobně jako u fosfitů a fosforamiditů, celkově lepší výsledky.[27]
Úspěšně byly také použity ligandy obsahující binaftylové struktury MonoPHOS[28] a spirocykly SiPHOS.[29]
Tyto monodentátní ligandy lze navzájem kombinovat a dosáhnout tak ještě lepší enantioselektivity;[30] to u difosfinových ligandů nelze.[27]



Chirální difosfinové ligandy
editovatDifosfinové ligandy se používají častěji než monofosfinové. Patří sem první ligand s vysokou selektivitou (DIOP), první ligand použitý v průmyslové asymetrické syntéze (DIPAMP)[31][32][3] a nejlepší známý chirální ligand (BINAP).[2] Chirální difosfinové ligandy se nyní používají zcela běžně.

Chirální P,N a P,O ligandy
editovat

Použití P,N ligandů při asymetrických hydrogenacích má původ u C2-symetrických bisoxazolinových ligandů.[33] Tyto ligandy byly ovšem brzy překonány monooxazolinovými ligandy, které postrádají C2 symetrii, což ale nemá vliv na účinnost asymetrické katalýzy.[34] P,N ligandy obsahují nechirální dusíkatý heterocykly, které jsou funkcionalizované skupinami obsahujícími fosfor; přesné vlastnosti heterocyklu a fosforového centra se ovšem mohou značně lišit. Žádná konkrétní skupina není dostatečně účinná u velkého počtu substrátů, i když byly zavedeny určité struktury (například fosfinooxazoliny, nebolii PHOX).[14][34][35] U úzce definované skupiny substrátu byla navíc při použití komplexů kovů s chirálními P,N ligandy téměř úplná přeměna a selektivita u jinak obtížně zpracovatelných systémů.[36] Některé komplexy odvozené od chelatujících P-O ligandů ukázaly dobré výsledky v hydrogenacích α,β-nenasycených ketonů a esterů.[37]
NHC ligandy
editovat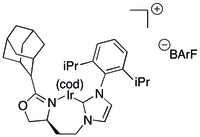
Ligandy obsahující jednoduché N-heterocyklické karbenové skupiny jsou pro asymetrické hydrogenace nevhodné.
Některé C,N ligandy vytvářejí s NHC a chirálními oxazoliny chelatující ligandy.[38][39] NHC ligandy prvního typu se získávají reakcemi NHC s oxazoliny.[38][39] NHC katalyzátory obsahující objemné sedmičlenné iridiové metalocykly byly v 80. a 90. letech 20. století použity na katalytické hydrogenace nefunkcionalizovaných alkenů[38] a vinyletheralkoholů s vysokými enantiomerními přebytky.[40] Stejný systém se také uplatnil při syntéze řady aldolových,[41] vicinálně dimethylových[42] a deoxypolyketidových sloučenin,[43] dokonce též na samotné deoxypolyketidy.[44]
C2-symetrické NHC se ukázaly jako velmi vhodné ligandy pro asymetrické hydrogenace.[45]
Acyklické substráty
editovatNenasycené acyklické substráty (alkeny, ketony, enaminy a iminy) jsou nejběžnějšími prochirálními substráty; substrát, které se nejvíce hodí k asymetrickým hydrogenacím často mají polární funkční skupiny v sousedství místa, jež má být hydrogenováno. Jestliže tyto skupiny nejsou přítomny, tak bývají enantiomerní přebytky obvykle nízké. V případě nefunkcionalizovaných alkenů jsou dobrými katalyzátory komplexy iridia s P,N ligandy. Využitelnost těchto katalyzátorů je neobvykle úzká; v důsledku toho bylo zvlášť zkoumáno a řešeno několik katalytických problémů, například u 1,1-disubstituovaných, 1,2-diaryltrisubstituovaných, 1,1,2-trialkylovaných a tetrasubstituovaných alkenů,[46][47]; i v rámci těchto skupin odchylky, pro které jsou nejvhodnější jiná řešení.[48]


Na rozdíl od alkenů probíhají asymetrické hydrogenace enaminů nejlépe za přítomnosti difosfinových ligandů; výborné výsledky byly dosaženy u systémů založených na iridiu i rhodiu. I u nejlepších z nich se ale vyskytovaly nízké enantiomerní přebytky a nedostatečná všeobecnost. Určité enaminy odvozené od pyrrolidinů a aromatických ketonů lze asymetricky hydrogenovat pomocí kationtových rhodných fosfonitových katalyzátorů a systémy založené na I2 a kyselině octové mají běžně přebytky kolem 90 %, potenciálně až 99,9 %.[49] Podobný systém využívající jednomocné iridium a fosforamidátový ligand má velkou účinnost u asymetrických hydrogenací pyrrolidinových enaminů s dvojnými vazbami uvnitř cyklů (dihydropyrrolů).[50] V obou případech dochází k významnému poklesu enantioselektivity při navýšení počtu atomů v kruhu z pěti na šest.
Iminy a ketony
editovat
Ketony a iminy jsou podobné funkční skupiny, i účinné postupy jejich asymetrické hydrogenace se podobají. Nejčastěji se zde používá Nojoriův katalytický systém tvořený rutheniem, chirálním difosfinem a diaminem.[51] Může být použit s mnoha různými fosfiny a aminy (amin nemusí nutně být chirální) a lze jej snadno upravit podle substrátu, dosahuje se přitom enantiomerních přebytků přes 90 %.[52][53]
U karbonylových a iminových substrátů se mohou společně vyskytovat η1 a η2 koordinace. U η1-vázaných substrátů se uhlík příjmající atom vodíku oddělí od katalyzátoru a není hydrogenován.[54]
Často se při asymetrických hydrogenacích ketonů a iminů používají také systémy založené na iridiu a P,N ligandech; například v případě benzylových aryliminů bývá používán P,N ligand SIPHOX ve spojení s iridným kationtovým komplexem a enantiomnerní přebytek bývá nad 90 %.[20] Jedním z nejúčinnějších vyvinutých katalyzátorů asymetrické hydrogenace ketonů, který má obratové číslo přibližně 4 550 000 a enantiomerní přebytky i 99,9 %, využívá jiný iridný systém s využitím příbuzného tridenátního ligandu.[15]

I přes podobnosti jsou tyto skupiny rozdílné, v mnoha oblastech se liší výrazně; například produkty asymetrické hydrogenace N-nefunkcionalizovaných iminů jsou primární aminy. Selektivní redukce iminů mohou být obtížné, protože se tyto sloučeniny vyskytují v rovnováze mezi iminovými a enaminovými tautomery a mezi (E) a (Z) izomery.[55] Jedním z řešení těchto potíží je použití ketiminů ve formě hydrochloridových solí, kde sterické vlastnosti alkylových či arylových skupin umožňují katalyzátoru rozlišit mezi enantiotopními stranami ketiminu.[56][57]
Aromatické substráty
editovatAsymetrické hydrogenace aromatických (obzvláště heteroaromatických) substrátů je předmětem mnoha výzkumů. Katalyzátory zde musejí překonat několik komplikací, jako jsou tendence velmi stabilních aromatických sloučenin odolávat hydrogenaci, možné koordinační reakce substrátů i produktů (které způsobují deaktivaci katalyzátoru) a značná rozmanitost substitucí, které mohou proběhnout na každém aromatickém kruhu.[58] Nejlépe se hydrogenují dusíkaté heterocykly, u nichž se aromatické jádro obvykle aktivuje protonací nebo další funkcionalizací na dusíku (většinou pomocí chránicí skupiny odtahující elektrony). Tyto postupy jsou méně využitelné u kyslíkatých a sirných heterocyklů, protože jsou slabšími zásadami i nukleofily; tím lze vysvětlit menší počet známých účinných způsobů jejich asymetrické hydrogenace.
Chinoliny, isochinoliny a chinoxaliny
editovatPro asymetrické hydrogenace 2-substituovaných chinolinů byly vyvinuty dva katalytické systémy; obecně mají výtěžnosti kolem 80 % a enantiomerní přebytky 90 %. První z nich se skládá z iridné sloučeniny, chirálního fosfinu aI2.[59] Prvním chirálním fosfinem použitým v tomto systému byl MeOBiPhep, nové systémy používají fosfiny (nebo podobné ligandy) vyznačující se lepší stabilitou na vzduchu,[60] recyklovatelností,[60] snadnou přípravou,[61] a nižším potřebným množstvím katalyzátoru.[19][62] K roku 2012 nebyl navržen žádný mechanismus, i když byla potvrzena jak nezbytnost použití I2, nebo náhrady halogenu, a možný vliv heteroaromatického dusíku na reaktivitu.[58]
Druhou možností je transferová hydrogenace katalyzovaná systémem založeným na Hantzschově esteru a chirální Brønstedově kyselině. Zde byl navržen mechanismus, ve kterém je isochinolin protonován v aktivačním kroku, poté redukován konjugovanou adicí hydridu z Hantzschova esteru.[63]

Většina vlastností asymetrické hydrogenace chinoxalinů úzce souvisí se strukturně podobnými chinoliny. Dobrých výsledků lze dosáhnout použitím katalytického systému IrI s fosfinitem a I2[64] nebo organokatalytického systému založeného na Hantzshově esteru,[65] přičemž oba se podobají systémům popsaným výše.
Pyridiny
editovatPyridiny jsou oproti jiným heteroaromatickým substrátům značně různorodou skupinou, protože pětice uhlíkových atomů umožňuje mnoho různých substitucí na kruhu.
Nejobecnější metoda asymetrické hydrogenace pyridinů je ve skutečnosti heterogenní, asymetrie u ní pochází z chirálního oxazolidinonu navázaného na pozici C2 pyridinu. Hydrogenacemi takto funkcionalizovaných pyridinů se provádí s odlišnými heterogenními kovovými katalyzátory, produkty jsou odpovídající piperidiny se substituenty na C3, C4 a C5, z nichž mají všechny cis geometrii, dosahuje se přitom vysokých výtěžností a výborných enantioselektivit. Oxazolidinony také mohou být hydrogenovány asenantioselektivně.[66]

Metody zaměřené přímo na 2-substituované pyridiny mohou zahrnovat asymetrické systémy vyvinuté pro podobné substraáty, jako jsou 2-substituované chinoliny a chinoxaliny, například systém IrI, chirálního fosfinu a I2 je vhodný pro asymetrické hydrogenace aktivovaných alkylovaných 2-pyridiniových sloučenin[67] nebo některých pyridinů spojených s cyklohexanony.[68] Podobně mohou chirální Brønstedovy kyseliny spolu s Hantzshovými estery jako zdroji hydridu účinně katalyzovat hydrogenace určitých 2-alkylpyridinů přes dodatečnou aktivující substituci.[69]
Indoly
editovatAsymetrické hydrogenace indolů byly původně zaměřeny na N-chráněné deriváty, kde chránicí skupina může aktivovat heterocyklus vůči hydrogenaci a zároveň sloužit jako místo ke koordinaci kovu. V pozdějších pracích byly nalezeny postupy hydrogenace nechráněných indolů skrz aktivace Brønstedovými kyselinami.
V první studii popisující asymetrickou hydrogenaci indolů byly použity 2-N-acetylované indoly a při vysokých výtěžnostech se dosahovalo enantiomerních přebytků 87 až 95 %. U 3-substituovaných indolů byla úspěšnost nižší, protože spolu s hydrogenací indolu docházelo rovněž k hydrolýze chránicí skupiny.[70] Při použití N-tosylových chránicích skupin byla hydrolýza znemožněna a mohly být s vysokými enantiomerními přebytky hydrogenovány jak 2-, tak i 3-substituované indoly.[71][72] U obou reakcí ovšem bylo k odstranění N-acetylových a N-tosylových skupin potřeba použít tvrdé reakční podmínky nevhodné pro složitější substráty. Tento problém byl vyřešen pomocí snadno oddělitelné N-Boc skupiny a byly brzy nalezeny velmi účinné postupy hydrogenace těchto indolů (jak 2-, tak i 3-substituovaných).[73][74]

I přes tyto pokroky v hydrogenacích chráněných indolů bylo dosaženo zjednodušení i u postupů nevyužívajících chránicí skupiny, a to katalytickými systémy využívajícími Brønstedovy kyseliny k aktivaci indolů. Původní systém obsahoval Pd(TFA)2/H8-BINAP k enantioselektivní cis-hydrogenaci 2,3- a 2-substituovaných indolů s dobrými výtěžnostmi a výbornými enantiomerními přebytky. Podobný proces, kde byly v jediné nádobě použity Friedelova–Craftsova alkylace a asymetrická hydrogenace, umožnil asymetrickou přípravu 2,3-substituovaných indolinů z 2-substituovaných indolů s podobnými výsledky.[75][76]

Jsou též známy postupy pro organokatalytické metodou asymetrické hydrogenace 2,3-substituovaných indolů s využitím chirálních Lewisových zásad, i když enantiomerní přebytky nebývají tak vysoké jako u hydrogenací katalyzovaných kovy.[75]
Pyrroly
editovatDosažení úplné přeměny pyrrolů na pyrrolidiny asymetrickou hydrogenací je náročné, protože často dochází jen k částečné hydrogenaci.[77][78] Úplná enantioselektivní redukce je možná, přičemž výsledek závisí na použitém substrátu a konkrétní metodě.
Asymetrické hydrogenace 2,3,5-substituovaných pyrrolů bylo dosaženo díky poznatku, že substráty vykazují stejné substituční vzorce jako 2-substituované indoly, a asymetrický hydrogenační systém účinný u jednoho typu může být dobře použitelný i u druhého. Následoval systém obsahující jednomocné ruthenium, fosfin a amin, použitelný na 2,3,5-substituované N-Boc pyrroly, jehož použití může vést, v závislosti na vlastnostech substituentů na pyrrolu, ke vzniku dihydro- i tetrahydropyrrolů (pyrrolidinů). Při substituci fenylovými skupinami se tvoří dihydropyrroly s výtěžností přes 96 % a velmi dobrou enantioselektivitou. Plně hydrogenované cis-dihydropyrroly se dají připravit diastereoselektivní heterogenní hydrogenací. Alkylové substituenty mohou vést k dihydro- i tetrahydropyrrolům, výtěžnosti (>70 %) a enantioselektivity (běžně >90 %) zůstávají vysoké. Regioselektivita je v obou případech řízena sterickými jevy, přednostně se hydrogenuje méně substituovaná dvojná vazba.[77]

Nechráněné 2,5-pyrroly se rovněž dají hydrogenovat pomocí Brønstedovy kyseliny, palladnatých katalyzátorů a chirálních fosfinů, produkty jsou 2,5-disubstituovsané 1-pyrroliny, s výtěžností zhruba 70 až 80 % a enantiomerním přebytkem 80 až 90 %.[78]
Kyslíkaté heterocykly
editovatAsymetrické hydrogenace furanů a benzofuranů se ukázaly jako obtížné.[79] Některé komplexy Ru-NHC katalyzují asymetrické hydrogenace benzofuranů[80] a furanů.[81] s dobrými mírami enantioindukce.

Sirné heterocykly
editovatPodobně jako u kyslíkatých heterocyklů je asymetrická hydrogenace sloučenin se sírou v nenasyceném pí systému omezena na thiofeny a benzothiofeny. Základní postup při asymetrické hydrogenaci těchto heterocyklů spočívá v ruthenatém katalyzátoru a chirálním C2-symetrikém N-heterocyklickém karbenu. Tento systém vykazuje výbornou selektivitu (s enantiomerním přebytkem přes 90 %) a diastereoselektivitu, jestliže má substrát kondenzovaná nebo přímo spojená fenylová jádra, ovšem v ostatních případech se vytváří pouze racemické produkty.[82]

Heterogenní katalýza
editovatKomerční využití nemá žádný heterogenní katalyzátor asymetrické hydrogenace.
První asymetrická hydrogenace byla provedena pomocí palladia na vrstvě hedvábí. Jako zdroje chirality byly použity alkaloidy z chinovníku (Cinchona).[83]

Jiným postupem, umožňujícím lépe řídit strukturní a elektronové vlastnosti aktivních katalytických míst, je imobilizace katalyzátorů vyvinutých pro homogenní katalýzu v heterogenních podmínkách. Nejčastějším způsobem provedení je kovalentní vazba katalyzátoru na polymer nebo jinou pevnou látku, i když lze také použít adsorbci katalyzátoru na povrch nebo výměnu iontů. Nevýhodou tohoto postupu je změna vlastností katalyzátoru a snížení enantioselektivity reakce. Aby se tomu zabránilo, tak se katalyzátory obvykle vážou přes jiné molekuly, které zvyšují vzdálenost od těchto povrchů a zvyšují účinnost katalyzátoru.[83]
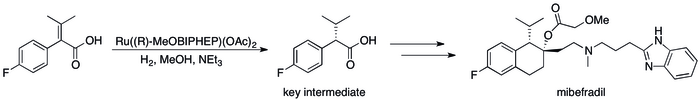
Použití v průmyslu
editovat-4_Ro_67-8867.svg)
Knowlesův výzkum asymetrické hydrogenace a jejího využití k výrobě L-Dopa[3] vyvolalo rozvoj průmyslových asymetrických hydrogenací. V roce 2001 zahrnovaly asymetrické hydrogenace 50 % vyrobených látek a 74 % enantioselektivních katalytických procesů v chemickém průmyslu, přičemž používání asymetrických katalýz bylo omezeno.[84]
Příklady využití asymetrické hydrogenace v průmyslu[85] může být řada postupů, kde bylo kinetické rozlišení nahrazeno vylepšenou účinností procesu; například se podařilo dosáhnout syntézy (S,S)-Ro 67-8867 s 53% výtěžností, což byl výrazný pokrok oproti 3,5 % dosahovaným syntézou na základě kinetického rozlišení.[86] Rocheova syntéza mibefradilu byla vylepšena nahrazením rozlišení asymetrickou hydrogenací, čímž se počet reakčních kroků snížil na tři a výtěžnost hlavního meziproduktu stoupla ze 70 % na 80 %.[87]
Reference
editovatV tomto článku byl použit překlad textu z článku Asymmetric hydrogenation na anglické Wikipedii.
- ↑ S. Akabori; S. Sakurai; Y. Izumi; Y. Fujii. An Asymmetric Catalyst. Nature. 1956, s. 323. DOI 10.1038/178323b0. PMID 13358737. Bibcode 1956Natur.178..323A.
- ↑ a b c R. Noyori. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture 2001). Advanced Synthesis & Catalysis. 2003, s. 15–41. DOI 10.1002/adsc.200390002.
- ↑ a b c W. S. Knowles. Asymmetric Hydrogenations (Nobel Lecture). Angewandte Chemie International Edition. 2002, s. 1998–2007. DOI 10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. PMID 19746594.
- ↑ A. Pfaltz. Asymmetric Catalysis Special Feature Part II: Design of chiral ligands for asymmetric catalysis: From C2-symmetric P,P- and N,N-ligands to sterically and electronically nonsymmetrical P,N-ligands. Proceedings of the National Academy of Sciences. 2004, s. 5723–5726. DOI 10.1073/pnas.0307152101. PMID 15069193. Bibcode 2004PNAS..101.5723P.
- ↑ a b I. D. Gridnev; T. Imamoto. On the Mechanism of Stereoselection in Rh-Catalyzed Asymmetric Hydrogenation: A General Approach for Predicting the Sense of Enantioselectivity. Accounts of Chemical Research. 2004, s. 633–644. DOI 10.1021/ar030156e. PMID 15379579.
- ↑ W. S. Knowles; M. J. Sabacky. Catalytic asymmetric hydrogenation employing a soluble, optically active, rhodium complex. Chemical Communications (London). 1968, s. 1445. DOI 10.1039/C19680001445.
- ↑ C. Pilkington; I. Lennon. The Application of Asymmetric Hydrogenation for the Manufacture of Pharmaceutical Intermediates:The Need for Catalyst Diversity. Synthesis. 2003, s. 1639. DOI 10.1055/s-2003-40871.
- ↑ A. Miyashita; A. Yasuda; H. Takaya; K. Toriumi; T. Ito; T. Souchi; R. Noyori. Synthesis of 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (BINAP), an atropisomeric chiral bis(triaryl)phosphine, and its use in the rhodium(I)-catalyzed asymmetric hydrogenation of α-(acylamino)acrylic acids. Journal of the American Chemical Society. 1980, s. 7932. DOI 10.1021/ja00547a020.
- ↑ R. Noyori; T. Ohkuma; M. Kitamura; H. Takaya; N. Sayo; H. Kumobayashi; S. Akutagawa. Asymmetric hydrogenation of β-keto carboxylic esters. A practical, purely chemical access to β-hydroxy esters in high enantiomeric purity. Journal of the American Chemical Society. 1987, s. 5856. DOI 10.1021/ja00253a051.
- ↑ Takeshi Ohkuma; Christian A. Sandoval; Rajagopal Srinivasan; Lin Quinghong; Wei Yinmao; Kilian Muñiz; Ryoji Noyori. Asymmetric Hydrogenation of tert-Alkyl Ketones. Journal of the American Chemical Society. 2005-06-01, s. 8288–8289. ISSN 0002-7863. DOI 10.1021/ja052071. PMID 15941254.
- ↑ T. Ohkuma; T. Hattori; H. Ooka; T. Inoue; R. Noyori. BINAP/1,4-Diamine−Ruthenium(II) Complexes for Efficient Asymmetric Hydrogenation of 1-Tetralones and Analogues. Organic Letters. 2004, s. 2681–2683. ISSN 0002-7863. DOI 10.1021/ol049157c. PMID 15281743.
- ↑ T. Ikariya; A. J. Blacker. Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Accounts of Chemical Research. 2007, s. 1300–1308. DOI 10.1021/ar700134q. PMID 17960897.
- ↑ T. L. Church; P. G. Andersson. Iridium catalysts for the asymmetric hydrogenation of olefins with nontraditional functional substituents. Coordination Chemistry Reviews. 2008, s. 513. DOI 10.1016/j.ccr.2007.09.015.
- ↑ a b A. Lightfoot; P. Schnider; A. Pfaltz. Enantioselective Hydrogenation of Olefins with Iridium-Phosphanodihydrooxazole Catalyst. Angewandte Chemie International Edition. 1998, s. 2897–2899. DOI 10.1002/(SICI)1521-3773(19981102)37:20<2897::AID-ANIE2897>3.0.CO;2-8. PMID 29711115.
- ↑ a b c J. H. Xie; X. Y. Liu; J. B. Xie; L. X. Wang; Q. L. Zhou. An Additional Coordination Group Leads to Extremely Efficient Chiral Iridium Catalysts for Asymmetric Hydrogenation of Ketones. Angewandte Chemie International Edition. 2011, s. 7329–7332. DOI 10.1002/anie.201102710. PMID 21751315.
- ↑ a b c A. Blankenstein, J. R. Blankenstein, R. Hilgraf, E. Hörmann, S. McIntyre, F. Menges, M. Schönleber, S. P. Smidt, B. Wüstenberg, N. Zimmermann. Iridium-Catalyzed Enantioselective Hydrogenation of Olefins. Advanced Synthesis & Catalysis. 2003, s. 33. DOI 10.1002/adsc.200390027.
- ↑ X. Cui; K. Burgess. Catalytic Homogeneous Asymmetric Hydrogenations of Largely Unfunctionalized Alkenes. Chemical Reviews. 2005, s. 3272–3296. DOI 10.1021/cr0500131. PMID 16159153.
- ↑ Y. Xu; D. M. P. Mingos; J. M. Brown. Crabtree's catalyst revisited; Ligand effects on stability and durability. Chemical Communications. 2008, s. 199–201. DOI 10.1039/B711979H. PMID 18092086.
- ↑ a b Z. J. Wang; G. J. Deng; Y. Li; Y. M. He; W. J. Tang; Q. H. Fan. Enantioselective Hydrogenation of Quinolines Catalyzed by Ir(BINAP)-Cored Dendrimers: Dramatic Enhancement of Catalytic Activity. Organic Letters. 2007, s. 1243–1246. DOI 10.1021/ol0631410. PMID 17328554.
- ↑ a b S. F. Zhu; J. B. Xie; Y. Z. Zhang; S. Li; Q. L. Zhou. Well-Defined Chiral Spiro Iridium/Phosphine−Oxazoline Cationic Complexes for Highly Enantioselective Hydrogenation of Imines at Ambient Pressure. Journal of the American Chemical Society. 2006, s. 12886–12891. DOI 10.1021/ja063444p. PMID 17002383.
- ↑ a b H. U. Blaser; B. T. Pugin; F. Spindler; A. Togni. Enantioselective imine hydrogenation with Ir diphosphine catalysts: Fighting deactivation. Comptes Rendus Chimie. 2002, s. 379. DOI 10.1016/S1631-0748(02)01391-7.
- ↑ S. Enthaler; K. Junge; M. Beller. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star?. Angewandte Chemie International Edition. 2008, s. 3317–3321. DOI 10.1002/anie.200800012. PMID 18412184.
- ↑ A. Mikhailine; A. J. Lough; R. H. Morris. Efficient Asymmetric Transfer Hydrogenation of Ketones Catalyzed by an Iron Complex Containing a P−N−N−P Tetradentate Ligand Formed by Template Synthesis. Journal of the American Chemical Society. 2009, s. 1394–1395. DOI 10.1021/ja809493h. PMID 19133772.
- ↑ J. F. Sonnenberg; N. Coombs; P. A. Dube; R. H. Morris. Iron Nanoparticles Catalyzing the Asymmetric Transfer Hydrogenation of Ketones. Journal of the American Chemical Society. 2012, s. 5893–5899. DOI 10.1021/ja211658t. PMID 22448656.
- ↑ J. K. Whitesell. C2 symmetry and asymmetric induction. Chemical Reviews. 1989, s. 1581–1590. DOI 10.1021/cr00097a012.
- ↑ W. S. Knowles; M. J. Sabacky; B. D. Vineyard. Catalytic asymmetric hydrogenation. Journal of the Chemical Society, Chemical Communications. 1972, s. 119–124. DOI 10.1039/C39720000010. PMID 4270504.
- ↑ a b T. Jerphagnon autor2 = J. L. Renaud; C. Bruneau. Chiral monodentate phosphorus ligands for rhodium-catalyzed asymmetric hydrogenation. Tetrahedron: Asymmetry. 2004, s. 2101. DOI 10.1016/j.tetasy.2004.04.037.
- ↑ M. Van Den Berg; A. J. Minnaard; E. P. Schudde; J. Van Esch; A. H. M. De Vries; J. G. De Vries; B. L. Feringa. Highly Enantioselective Rhodium-Catalyzed Hydrogenation with Monodentate Ligands. Journal of the American Chemical Society. 2000, s. 11539. Dostupné online. DOI 10.1021/ja002507f.
- ↑ Y. Fu; J. H. Xie; A. G. Hu; H. Zhou; L. X. Wang; Q. L. Zhou. Novel monodentate spiro phosphorus ligands for rhodium-catalyzed hydrogenation reactions. Chemical Communications. 2002, s. 480–481. DOI 10.1039/B109827F. PMID 12120551.
- ↑ M. T. Reetz; T. Sell; A. Meiswinkel; G. Mehler. A New Principle in Combinatorial Asymmetric Transition-Metal Catalysis: Mixtures of Chiral Monodentate P Ligands. Angewandte Chemie International Edition. 2003, s. 790–793. DOI 10.1002/anie.200390209. PMID 12596201.
- ↑ B. D. Vineyard; W. S. Knowles; M. J. Sabacky; G. L. Bachman; D. J. Weinkauff. Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. Journal of the American Chemical Society. 1977, s. 5946. DOI 10.1021/ja00460a018.
- ↑ W. S. Knowles; M. J. Sabacky; B. D. Vineyard; D. J. Weinkauff. Asymmetric hydrogenation with a complex of rhodium and a chiral bisphosphine. Journal of the American Chemical Society. 1975, s. 2567. DOI 10.1021/ja00842a058.
- ↑ MÜLLER, D.; UMBRICHT, G.; WEBER, B.; PFALTZ, A. C2-Symmetric 4,4',5,5'-Tetrahydrobi(oxazoles) and 4,4',5,5'-Tetrahydro-2,2'-methylenebis[oxazoles] as Chiral Ligands for Enantioselective Catalysis Preliminary Communication. Helvetica Chimica Acta. 1991, s. 232–240. DOI 10.1002/hlca.19910740123.
- ↑ a b HELMCHEN, G. N.; PFALTZ, A. PhosphinooxazolinesA New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis. Accounts of Chemical Research. 2000, s. 336–345. DOI 10.1021/ar9900865. PMID 10891051.
- ↑ FRANZKE, A.; PFALTZ, A. Zwitterionic Iridium Complexes with P,N-Ligands as Catalysts for the Asymmetric Hydrogenation of Alkenes. Chemistry: A European Journal. 2011, s. 4131–44. DOI 10.1002/chem.201003314. PMID 21381140.
- ↑ MAURER, F.; HUCH, V.; ULLRICH, A.; KAZMAIER, U. Development of Catalysts for the Stereoselective Hydrogenation of α,β-Unsaturated Ketones. The Journal of Organic Chemistry. 2012, s. 5139–5143. DOI 10.1021/jo300246c. PMID 22571628.
- ↑ RAGEOT, D.; WOODMANSEE, D. H.; PUGIN, B. T.; PFALTZ, A. Proline-Based P,O Ligand/Iridium Complexes as Highly Selective Catalysts: Asymmetric Hydrogenation of Trisubstituted Alkenes. Angewandte Chemie International Edition. 2011, s. 9598–601. DOI 10.1002/anie.201104105. PMID 21882320.
- ↑ a b c PERRY, M. C.; CUI, X.; POWELL, M. T.; HOU, D. R.; REIBENSPIES, J. H.; BURGESS, K. Optically Active Iridium Imidazol-2-ylidene-oxazoline Complexes: Preparation and Use in Asymmetric Hydrogenation of Arylalkenes. Journal of the American Chemical Society. 2003, s. 113–123. DOI 10.1021/ja028142b. PMID 12515512.
- ↑ a b NANCHEN, S.; PFALTZ, A. Synthesis and Application of Chiral N-Heterocyclic Carbene–Oxazoline Ligands: Iridium-Catalyzed Enantioselective Hydrogenation. Chemistry: A European Journal. 2006, s. 4550–8. DOI 10.1002/chem.200501500. PMID 16557626.
- ↑ ZHU, Y.; BURGESS, K. Iridium-Catalyzed Asymmetric Hydrogenation of Vinyl Ethers. Advanced Synthesis & Catalysis. 2008, s. 979. DOI 10.1002/adsc.200700546.
- ↑ ZHAO, J.; BURGESS, K. Aldol-Type Chirons from Asymmetric Hydrogenations of Trisubstituted Alkenes. Organic Letters. 2009, s. 2053–2056. DOI 10.1021/ol900308w. PMID 19368378.
- ↑ ZHAO, J.; BURGESS, K. Synthesis of Vicinal Dimethyl Chirons by Asymmetric Hydrogenation of Trisubstituted Alkenes. Journal of the American Chemical Society. 2009, s. 13236–13237. DOI 10.1021/ja905458n. PMID 19719102.
- ↑ ZHOU, J.; BURGESS, K. Α,ω-Functionalized 2,4-Dimethylpentane Dyads and 2,4,6-Trimethylheptane Triads through Asymmetric Hydrogenation. Angewandte Chemie International Edition. 2007, s. 1129–31. DOI 10.1002/anie.200603635. PMID 17200966.
- ↑ ZHOU, J.; ZHU, Y.; BURGESS, K. Synthesis of (S,R,R,S,R,S)-4,6,8,10,16,18- Hexamethyldocosane from Antitrogus parvulus via Diastereoselective Hydrogenations. Organic Letters. 2007, s. 1391–1393. DOI 10.1021/ol070298z. PMID 17338543.
- ↑ URBAN, S.; ORTEGA, N.; GLORIUS, F. Ligand-Controlled Highly Regioselective and Asymmetric Hydrogenation of Quinoxalines Catalyzed by Ruthenium N-Heterocyclic Carbene Complexes. Angewandte Chemie International Edition. 2011, s. 3803–6. DOI 10.1002/anie.201100008. PMID 21442699.
- ↑ PÀMIES, O.; ANDERSSON, P. G.; DIÉGUEZ, M. Asymmetric Hydrogenation of Minimally Functionalised Terminal Olefins: An Alternative Sustainable and Direct Strategy for Preparing Enantioenriched Hydrocarbons. Chemistry: A European Journal. 2010, s. 14232–40. DOI 10.1002/chem.201001909. PMID 21140401.
- ↑ WOODMANSEE, D. H.; PFALTZ, A. Asymmetric hydrogenation of alkenes lacking coordinating groups. Chemical Communications. 2011, s. 7912–7916. DOI 10.1039/c1cc11430a. PMID 21556431.
- ↑ MAZUELA, J.; VERENDEL, J. J.; COLL, M.; SCHÄFFNER, B. N.; BÖRNER, A.; ANDERSSON, P. G.; PÀMIES, O. Iridium Phosphite−Oxazoline Catalysts for the Highly Enantioselective Hydrogenation of Terminal Alkenes. Journal of the American Chemical Society. 2009, s. 12344–12353. DOI 10.1021/ja904152r. PMID 19658416.
- ↑ HOU, G. H.; XIE, J. H.; WANG, L. X.; ZHOU, Q. L. Highly Efficient Rh(I)-Catalyzed Asymmetric Hydrogenation of Enamines Using Monodente Spiro Phosphonite Ligands. Journal of the American Chemical Society. 2006, s. 11774–11775. DOI 10.1021/ja0644778. PMID 16953614.
- ↑ HOU, G. H.; XIE, J. H.; YAN, P. C.; ZHOU, Q. L. Iridium-Catalyzed Asymmetric Hydrogenation of Cyclic Enamines. Journal of the American Chemical Society. 2009, s. 1366–1367. DOI 10.1021/ja808358r. PMID 19132836.
- ↑ OHKUMA, T.; OOKA, H.; HASHIGUCHI, S.; IKARIYA, T.; NOYORI, R. Practical Enantioselective Hydrogenation of Aromatic Ketones. Journal of the American Chemical Society. 1995, s. 2675. DOI 10.1021/ja00114a043.
- ↑ NOYORI, R.; OHKUMA, T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angewandte Chemie International Edition. 2001, s. 40–73. DOI 10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5. PMID 11169691.
- ↑ HEMS, W. P.; GROARKE, M.; ZANOTTI-GEROSA, A.; GRASA, G. A. [(Bisphosphine) Ru(II) Diamine] Complexes in Asymmetric Hydrogenation: Expanding the Scope of the Diamine Ligand. Accounts of Chemical Research. 2007, s. 1340–1347. DOI 10.1021/ar7000233. PMID 17576143.
- ↑ NOYORI, R.; YAMAKAWA, M.; HASHIGUCHI, S. Metal−Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds. The Journal of Organic Chemistry. 2001, s. 7931–7944. DOI 10.1021/jo010721w. PMID 11722188.
- ↑ YU, Z.; JIN, W.; JIANG, Q. Brønsted Acid Activation Strategy in Transition-Metal Catalyzed Asymmetric Hydrogenation of N-Unprotected Imines, Enamines, and N-Heteroaromatic Compounds. Angewandte Chemie International Edition. 2012, s. 6060–72. DOI 10.1002/anie.201200963. PMID 22577004.
- ↑ HOU, G.; GOSSELIN, F.; LI, W.; MCWILLIAMS, J. C.; SUN, Y.; WEISEL, M.; O'SHEA, P. D. Enantioselective Hydrogenation of N−H Imines. Journal of the American Chemical Society. 2009, s. 9882–9883. DOI 10.1021/ja903319r. PMID 19569686.
- ↑ HOU, G.; TAO, R.; SUN, Y.; ZHANG, X.; GOSSELIN, F. Iridium−Monodentate Phosphoramidite-Catalyzed Asymmetric Hydrogenation of Substituted Benzophenone N−H Imines. Journal of the American Chemical Society. 2010, s. 2124–2125. DOI 10.1021/ja909583s. PMID 20104899.
- ↑ a b ZHOU, Y. G. Asymmetric Hydrogenation of Heteroaromatic Compounds. Accounts of Chemical Research. 2007, s. 1357–1366. DOI 10.1021/ar700094b. PMID 17896823.
- ↑ WANG, W. B.; LU, S. M.; YANG, P. Y.; HAN, X. W.; ZHOU, Y. G. Highly Enantioselective Iridium-Catalyzed Hydrogenation of Heteroaromatic Compounds, Quinolines. Journal of the American Chemical Society. 2003, s. 10536–10537. DOI 10.1021/ja0353762. PMID 12940733.
- ↑ a b XU, L.; LAM, K. H.; JI, J.; WU, J.; FAN, Q. H.; LO, W. H.; CHAN, A. S. C. Air-stable Ir-(P-Phos) complex for highly enantioselective hydrogenation of quinolines and their immobilization in poly(ethylene glycol) dimethyl ether (DMPEG). Chemical Communications. 2005, s. 1390–2. DOI 10.1039/B416397D. PMID 15756313.
- ↑ LAM, K. H.; XU, L.; FENG, L.; FAN, Q. H.; LAM, F. L.; LO, W. H.; CHAN, A. S. C. Highly Enantioselective Iridium-Catalyzed Hydrogenation of Quinoline Derivatives Using Chiral Phosphinite H8-BINAPO. Advanced Synthesis & Catalysis. 2005, s. 1755. DOI 10.1002/adsc.200505130.
- ↑ QIU, L.; KWONG, F. Y.; WU, J.; LAM, W. H.; CHAN, S.; YU, W. Y.; LI, Y. M. A New Class of Versatile Chiral-Bridged Atropisomeric Diphosphine Ligands: Remarkably Efficient Ligand Syntheses and Their Applications in Highly Enantioselective Hydrogenation Reactions. Journal of the American Chemical Society. 2006, s. 5955–5965. DOI 10.1021/ja0602694. PMID 16637664.
- ↑ Rueping; ANTONCHICK, A.; THEISSMANN, T. A highly enantioselective Brønsted acid catalyzed cascade reaction: organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids. Angewandte Chemie International Edition in English. 2006, s. 3683–3686. DOI 10.1002/anie.200600191. PMID 16639754.
- ↑ TANG, W.; XU, L.; FAN, Q. H.; WANG, J.; FAN, B.; ZHOU, Z.; LAM, K. H. Asymmetric Hydrogenation of Quinoxalines with Diphosphinite Ligands: A Practical Synthesis of Enantioenriched, Substituted Tetrahydroquinoxalines. Angewandte Chemie International Edition. 2009, s. 9135–9138. DOI 10.1002/anie.200904518. PMID 19876991.
- ↑ RUEPING, M.; TATO, F.; SCHOEPKE, F. R. The First General, Efficient and Highly Enantioselective Reduction of Quinoxalines and Quinoxalinones. Chemistry: A European Journal. 2010, s. 2688–91. DOI 10.1002/chem.200902907. PMID 20140920.
- ↑ GLORIUS, F.; SPIELKAMP, N.; HOLLE, S.; GODDARD, R.; LEHMANN, C. W. Efficient Asymmetric Hydrogenation of Pyridines. Angewandte Chemie International Edition. 2004, s. 2850–2. DOI 10.1002/anie.200453942. PMID 15150766.
- ↑ YE, Z. S.; CHEN, M. W.; CHEN, Q. A.; SHI, L.; DUAN, Y.; ZHOU, Y. G. Iridium-Catalyzed Asymmetric Hydrogenation of Pyridinium Salts. Angewandte Chemie International Edition. 2012, s. 10181–4. DOI 10.1002/anie.201205187. PMID 22969060.
- ↑ TANG, W. J.; TAN, J.; XU, L. J.; LAM, K. H.; FAN, Q. H.; CHAN, A. S. C. Highly Enantioselective Hydrogenation of Quinoline and Pyridine Derivatives with Iridium-(P-Phos) Catalyst. Advanced Synthesis & Catalysis. 2010, s. 1055. DOI 10.1002/adsc.200900870.
- ↑ RUEPING, M.; ANTONCHICK, A. P. Organocatalytic Enantioselective Reduction of Pyridines. Angewandte Chemie International Edition. 2007, s. 4562–5. DOI 10.1002/anie.200701158. PMID 17492817.
- ↑ KUWANO, R.; SATO, K.; KUROKAWA, T.; KARUBE, D.; ITO, Y. Catalytic Asymmetric Hydrogenation of Heteroaromatic Compounds, Indoles. Journal of the American Chemical Society. 2000, s. 7614. DOI 10.1021/ja001271c.
- ↑ KUWANO, R.; KANEDA, K.; ITO, T.; SATO, K.; KUROKAWA, T.; ITO, Y. Highly Enantioselective Synthesis of Chiral 3-Substituted Indolines by Catalytic Asymmetric Hydrogenation of Indoles. Organic Letters. 2004, s. 2213–2215. DOI 10.1021/ol049317k. PMID 15200323.
- ↑ KUWANO, R.; KASHIWABARA, M.; SATO, K.; ITO, T.; KANEDA, K.; ITO, Y. Catalytic asymmetric hydrogenation of indoles using a rhodium complex with a chiral bisphosphine ligand PhTRAP. Tetrahedron: Asymmetry. 2006, s. 521. DOI 10.1016/j.tetasy.2006.01.016.
- ↑ KUWANO, R.; KASHIWABARA, M. Ruthenium-Catalyzed Asymmetric Hydrogenation of N-Boc-Indoles. Organic Letters. 2006, s. 2653–2655. DOI 10.1021/ol061039x. PMID 16737337.
- ↑ BAEZA, A.; PFALTZ, A. Iridium-Catalyzed Asymmetric Hydrogenation of N-Protected Indoles. Chemistry: A European Journal. 2010, s. 2036–9. DOI 10.1002/chem.200903105. PMID 20104554.
- ↑ a b XIAO, Y. C.; WANG, C.; YAO, Y.; SUN, J.; CHEN, Y. C. Direct Asymmetric Hydrosilylation of Indoles: Combined Lewis Base and Brønsted Acid Activation. Angewandte Chemie International Edition. 2011, s. 10661–4. DOI 10.1002/anie.201105341. PMID 21932274.
- ↑ DUAN, Y.; CHEN, M. W.; YE, Z. S.; WANG, D. S.; CHEN, Q. A.; ZHOU, Y. G. An Enantioselective Approach to 2,3-Disubstituted Indolines through Consecutive Brønsted Acid/Pd-Complex-Promoted Tandem Reactions. Chemistry: A European Journal. 2011, s. 7193–7. DOI 10.1002/chem.201100576. PMID 21567504.
- ↑ a b KUWANO, R.; KASHIWABARA, M.; OHSUMI, M.; KUSANO, H. Catalytic Asymmetric Hydrogenation of 2,3,5-Trisubstituted Pyrroles. Journal of the American Chemical Society. 2008, s. 808–809. DOI 10.1021/ja7102422. PMID 18154340.
- ↑ a b WANG, D. S.; YE, Z. S.; CHEN, Q. A.; ZHOU, Y. G.; YU, C. B.; FAN, H. J.; DUAN, Y. Highly Enantioselective Partial Hydrogenation of Simple Pyrroles: A Facile Access to Chiral 1-Pyrrolines. Journal of the American Chemical Society. 2011, s. 8866–8869. DOI 10.1021/ja203190t. PMID 21591641.
- ↑ WANG, D. S.; CHEN, Q. A.; LU, S. M.; ZHOU, Y. G. Asymmetric Hydrogenation of Heteroarenes and Arenes. Chemical Reviews. 2012, s. 2557–2590. DOI 10.1021/cr200328h. PMID 22098109.
- ↑ ORTEGA, Nuria; URBAN, Slawomir; BEIRING, Bernhard; GLORIUS, Frank. Ruthenium NHC Catalyzed Highly Asymmetric Hydrogenation of Benzofurans. Angewandte Chemie International Edition. 2012, s. 1710–3. DOI 10.1002/anie.201107811. PMID 22311814.
- ↑ WYSOCKI, Jędrzej; ORTEGA, Nuria; GLORIUS, Frank. Asymmetric Hydrogenation of Disubstituted Furans. Angewandte Chemie International Edition. 2014, s. 8751–5. DOI 10.1002/anie.201310985. PMID 24554623.
- ↑ URBAN, S.; BEIRING, B.; ORTEGA, N.; PAUL, D.; GLORIUS, F. Asymmetric Hydrogenation of Thiophenes and Benzothiophenes. Journal of the American Chemical Society. 2012, s. 15241–15244. DOI 10.1021/ja306622y. PMID 22934527.
- ↑ a b HEITBAUM, M.; GLORIUS, F.; ESCHER, I. Asymmetric Heterogeneous Catalysis. Angewandte Chemie International Edition. 2006, s. 4732–62. DOI 10.1002/anie.200504212. PMID 16802397.
- ↑ BLASER, H. U.; SPINDLER, F.; STUDER, M. Enantioselective catalysis in fine chemicals production. Applied Catalysis A: General. 2001, s. 119–143. DOI 10.1016/S0926-860X(01)00801-8. PMID 12613584.
- ↑ DUB, Pavel A.; GORDON, John C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nature Reviews Chemistry. 2018, s. 396–408. DOI 10.1038/s41570-018-0049-z.
- ↑ Asymmetric Catalysis on Industrial Scale. Redakce Blaser Hans-Ulrich. Weinheim: Wiley-VCH, 2010. ISBN 978-3-527-63063-9. DOI 10.1002/9783527630639. S. 13–16.
- ↑ Comprehensive Asymmetric Catalysis. Redakce Jacobsen E. N.. Berlin; New York: Springer, 1999. ISBN 978-3-540-64336-4. S. 1443–1445.